
Purpose of This Guideline
Date of current publication: June 30, 2023
Lead author: Steven M. Fine, MD, PhD
Writing group: Rona M. Vail, MD; Joseph P. McGowan, MD, FACP, FIDSA; Samuel T. Merrick, MD; Asa E. Radix, MD, MPH, PhD; Jessica Rodrigues, MS; Charles J. Gonzalez, MD; Christopher J. Hoffmann, MD, MPH
Committee: Medical Care Criteria Committee
Date of original publication: July 13, 2016
Clinical trials as well as ample anecdotal evidence support better antiretroviral therapy (ART) outcomes when HIV resistance testing is performed Palella, et al. 2009; Baxter, et al. 2000. This guideline was developed by the New York State Department of Health AIDS Institute (NYSDOH AI) as a resource for clinicians in New York State who provide care for patients with HIV. The guideline includes evidence-based recommendations for using HIV drug resistance testing to help improve ART outcomes. Toward that end, the goals of this guideline are to:
- Assist clinicians in determining when to order HIV drug resistance testing
- Inform clinicians about the different types, benefits, and limitations of currently available resistance assays
- Assist clinicians in choosing resistance testing to improve treatment outcomes
Note on “experienced” HIV care providers: The NYSDOH AI Clinical Guidelines Program defines an “experienced HIV care provider” as a practitioner who has been accorded HIV Specialist status by the American Academy of HIV Medicine. Nurse practitioners (NPs) and licensed midwives who provide clinical care to individuals with HIV in collaboration with a physician may be considered experienced HIV care providers if all other practice agreements are met; NPs with more than 3,600 hours of qualifying experience do not require collaboration with a physician (8 NYCRR 79-5:1; 10 NYCRR 85.36; 8 NYCRR 139-6900). Physician assistants who provide clinical care to individuals with HIV under the supervision of an HIV Specialist physician may also be considered experienced HIV care providers (10 NYCRR 94.2).
Determining HIV Drug Resistance
| RECOMMENDATIONS |
Determining HIV Drug Resistance
|
Abbreviations: ART, antiretroviral therapy; CAB/RPV LA, injectable long-acting cabotegravir and rilpivirine. Notes:
|
Interpreting HIV resistance assays can be challenging but is necessary to craft a reliably suppressive ART regimen. HIV replicates via reverse transcriptase, which lacks proofreading capacity and is susceptible to mutations during viral replication (i.e., changes in its genetic sequence). Random mutations coupled with the selective pressure of subtherapeutic drug levels can lead to selection of drug-resistant HIV strains. Once established, drug-resistant HIV strains may continue to replicate even in patients who are adherent to their ART regimens. Thus, if a patient’s HIV viral load remains detectable and they are known to be adherent to their ART regimen, resistance testing should be performed Kantor and Gupta 2023; Wood and Stekler 2022.
| KEY POINT |
|
The most commonly used ART drugs are targeted to inhibit the activity of 3 specific viral enzymes: protease, reverse transcriptase, and integrase. Mutations have been identified that interfere with the ability of 1 or more ART agents to inhibit viral replication, thus rendering the virus resistant to the drug(s). HIV resistance mutations for less commonly used ART drugs that target fusion, viral entry, and capsid formation have also been identified.
As new ART drugs are developed and knowledge about the clinical significance of resistance mutations evolves, consulting the following resources for information on drug resistance mutations and resistance testing may be useful:
- Stanford University HIV Drug Resistance Database
- International Antiviral Society-USA 2022 Update of the Drug Resistance Mutations in HIV-1
- HIV Resistance Response Database Initiative
- Los Alamos National Laboratory HIV Databases
- HIV French Resistance Database
HIV resistance testing is generally accomplished through genotyping, which directly sequences the HIV genome to detect mutations. Phenotyping, which measures HIV replication in the presence of drug, may also be available, but genotyping is generally preferred. Single-copy and deep sequencing technologies that analyze proviral DNA are also available in special circumstances to detect mutations present at very low levels or in virally suppressed patients. Each of these options for HIV drug resistance testing is discussed in the guideline section Genotypic and Phenotypic Resistance Assays.
Insurance coverage of drug resistance testing: In New York State, health plans may impose annual limits on the number of resistance tests that can be performed for a given patient, and some plans may require prior authorization. Refer to the patient’s specific insurance plan regarding frequency, annual limits, and whether prior authorization is required for any genotypic and phenotypic HIV resistance tests.
Genotypic and Phenotypic Resistance Assays
Genotyping
Sanger and next-generation sequencing: Genotypic resistance assays directly sequence the protein coding regions of genes for viral reverse transcriptase, protease, and integrase. The mutations identified via genotyping are compared with a list of mutations that are known to confer resistance to current antiretroviral medications (ARVs), generating a list of ARVs that are likely to remain active against virus versus those to which resistance is likely.
When interpreting resistance test results, it is important to include mutations from all prior resistance tests whenever available because these mutations are thought to be cumulative in most cases. The resources noted in the guideline section Determining HIV Drug Resistance may be helpful when interpreting genotypic resistance testing results, but consultation with an expert HIV care provider is also advised.
Currently, there are 2 methods for sequencing for genotypic drug resistance:
- Standard genotype sequencing (Sanger or population sequencing) derives a consensus sequence for each gene in the sample and is useful for detecting mutations that are dominant or are present at high levels in the sample. Sanger sequencing is still used at most sites.
- Next-generation sequencing (NGS) FDA 2019, which is beginning to replace standard genotyping technology at some sites, allows for simultaneous sequencing of thousands of individual genomes and can detect mutations at far lower copy numbers than Sanger sequencing. NGS may detect low copy number mutants in patients who have stopped antiretroviral therapy (ART) for some time and in whom the majority of replicating HIV has been replaced by wild-type sequences.
Genotypic resistance assays generally require that an individual have an HIV RNA level (viral load) of >500 copies/mL, but individual laboratories may have their own minimum viral load requirements.
Proviral DNA-based (archive) genotypic assays: NGS technology can also be used to sequence HIV proviral DNA, which is nonreplicating, in patients who may wish to change their suppressive ART regimen. With these assays, integrated proviral DNA is extracted from within the cell and the coding sequences for reverse transcriptase, protease, and integrase are sequenced as they are in the standard genotypic assays, to predict whether a virus derived from these proviral sequences would be resistant to ARVs Chu, et al. 2022.
For patients whose previous ART regimens failed and whose viral load is currently undetectable, and for whom past genotype test results are not available, proviral DNA-based genotypic assays may detect “archived” mutations; these assays are often referred to as “archive genotypes.” Archive genotypes may be useful for patients with a complex ART history or when previous resistance test results are unavailable, but the clinical significance of archived mutations is not yet fully understood.
One potential use of archive genotypes is for patients who are currently on a suppressive oral ART regimen but are considering switching to CAB/RPV LA. In this situation, the archive genotype might identify non-nucleoside reverse transcriptase inhibitor resistance-associated mutations, which are known to increase the risk of treatment failure.
Because the availability of HIV genotypic resistance testing is evolving, it is best to check with local laboratories to see which tests are currently available and for specific sample requirements.
Phenotyping
Although still available, phenotypic assays generally do not add to the information provided by currently used genotypic assays. If a patient has experienced multiple regimen failures and has a large number of ARV resistance mutations, which is rare, a phenotypic assay may be used to help devise an effective ART regimen. A phenotypic assay provides a direct measure of drug resistance and is analogous to antibiotic-susceptibility testing of bacteria. Currently available phenotypic assays use recombinant DNA methods to measure the ability of a patient’s virus to replicate in the presence of a drug. Therefore, results from a phenotypic test include the net effect of multiple resistance mutations.
In the phenotypic assay, HIV RNA is isolated from plasma and converted into cDNA, and the relevant region is amplified by polymerase chain reaction (PCR). This amplified material is inserted into a recombinant virus system whereby the susceptibility to different drugs can be tested. The result from the phenotypic assay is a value that defines the concentration of the drug required to reduce growth of the virus by 50% (IC50). The IC50 of the patient’s virus is compared with the IC50 of a drug-sensitive (wild-type) reference virus, and the fold change is defined. If the IC50 of an individual’s virus is greater than that of the reference virus for a particular drug, then the individual’s virus has decreased sensitivity to the drug. The relative fold change helps determine whether the drug should still be included in the ART regimen or whether it should be removed entirely. Monogram Biosciences offers phenotypic resistance testing through clinical laboratories with the PhenoSense assay. A phenotypic test for resistance to enfuvirtide that may help predict the drug’s activity is available through Labcorp-Monogram Biosciences. Phenotypic assays have a minimum viral load requirement of 500 to 1,000 copies/mL and results are generally available in 3 to 5 weeks.
Phenotypic assays are more technically complex, labor-intensive, and expensive than genotypic assays.
Technical Limitations of Genotypic and Phenotypic Assays
In addition to the minimum viral load requirements needed for amplification (generally at least 500 to 1,000 copies/mL) in standard genotypic or phenotypic RNA-based resistance assays, all resistance assays, including the DNA-based genotype, are limited by sampling bias. Acute HIV infection is often established by a single progenitor virion Cohen, et al. 2011, whereas in established HIV infection, HIV exists as a virus population comprising multiple genomic variants. Genotypic and phenotypic resistance assays are each more likely to detect the common viral variants and fail to identify the minor variants. Similarly, standard genotypic and phenotypic resistance testing performed on plasma specimens will not detect noncirculating or archived virus containing resistance-associated mutations (i.e., nonreplicating virus present within cells as proviral DNA). If ART is interrupted, the selective pressure from the ART is removed and generally favors replication of wild-type HIV strains, which eventually will predominate. When this occurs, the RNA-based genotypic and phenotypic resistance assays may fail to detect low-level ART-resistant virus. A DNA-based proviral assay may have utility in these circumstances; see the NYSDOH AI guideline Second-Line ART After Treatment Failure or for Regimen Simplification > Identifying and Managing Virologic Failure for more information. For these reasons, all previous genotype and phenotype resistance testing results, along with the patient’s ART medication history, should be retained, and this information should be combined and used in constructing a subsequent ART regimen. Once resistance develops, it can be expected to persist indefinitely to that specific drug in archived form.
Replicative Capacity
Replicative capacity information may be provided as an adjunct to phenotypic or combination genotypic-phenotypic resistance assays. The relative replicative capacity of the virus in a patient is calculated as the ratio of patient-derived sequences to wild-type sequences. A ratio of less than 1 reflects a reduced replicative capacity compared with that of the wild-type control. The full clinical value of this adjunctive information remains under investigation, and it has no clear clinical value at this time.
Coreceptor Tropism Assay
CCR5 and CXCR4 tropism: HIV-1 binds to a chemokine receptor, either CCR5 or CXCR4, as a coreceptor to enter CD4 cells. Viruses that use CCR5 are referred to as CCR5 (or R5) tropic and those that use CXCR4 as CXCR4 (or X4) tropic. The antiretroviral drug maraviroc blocks HIV from binding to CCR5. Therefore, if use of maraviroc is being considered, it is essential to determine that no CXCR4-tropic virus is present. Both genotypic and phenotypic assays measure CCR5 and CXCR4 tropism. Generally, the choice of which assay to use will be determined by which assay is locally available (see discussion below). CCR5-tropic virus predominates early in HIV infection, whereas CXCR4-tropic virus is often present in late-stage disease, and virus that uses CCR5 may be preferentially transmitted compared with CXCR4 variants. The majority of individuals with acute or recent HIV infection, including perinatally infected children, have CCR5-tropic virus.
In patients with chronic HIV infection, a population of mixed CCR5- and CXCR4-tropic viruses, as well as dual-tropic (able to use either CCR5 or CXCR4 as coreceptor) viruses, may be detected. The tropism of these viral populations is often referred to as dual/mixed or D/M HIV.
Resistance to CCR5 coreceptor antagonists develops by 2 unrelated mechanisms. The first, and most common, occurs when a patient’s viral population shifts its coreceptor usage from CCR5 to CXCR4. In this scenario, both CXCR4- and dual-tropic virus will be resistant to CCR5 antagonists. The second, and more rare, occurs when a virus develops the ability to continue to use CCR5 as its coreceptor despite the binding of the CCR5 antagonist. This latter type of resistance will be detected using phenotypic resistance assays.
Coreceptor tropism testing: Coreceptor tropism should be tested whenever the use of a CCR5 entry inhibitor (e.g., maraviroc) is being considered or when a patient’s CCR5 antagonist-containing regimen is failing despite excellent adherence. Coreceptor testing can either be phenotypic or genotypic, but phenotypic tropism testing is most commonly used.
The phenotypic coreceptor tropism assay (Trofile) measures inhibition of viral replication by CCR5 antagonists in vitro. If the HIV viral load is undetectable, a DNA tropism assay (Trofile DNA) can be used instead. In the DNA tropism assay, proviral DNA is first extracted from peripheral blood mononuclear cells (PBMCs) and then used in the phenotypic assay to predict whether CXCR4- or dual-tropic sequences are present.
Genotypic coreceptor tropism assays predict tropism based on sequencing of the V3 region of the HIV env gene Vandekerckhove, et al. 2011; McGovern, et al. 2010. A standard genotyping assay can be performed if the HIV viral load is >1,000 copies/mL, and a proviral DNA genotype is used if the viral load is undetectable. These assays are currently available through Quest Diagnostics. Studies of genotypic assays suggest a lower sensitivity than phenotypic assays for finding CXCR4- or dual-tropic virus, but clinical trials suggest these assays are good at predicting response to maraviroc. No trial has compared phenotypic and genotypic coreceptor tropism assays head to head, so the best use of genotypic assays is still unclear. At this time, most tropism testing in the United States is performed using phenotypic assays.
When use of a CCR5 antagonist (i.e., maraviroc) is being considered or a CCR5 antagonist-containing ART regimen is failing, either the standard phenotypic (if viral load is ≥1,000 copies/mL) or genotypic (if viral load is <1,000 copies/mL) coreceptor tropism assay should be used.
All Recommendations
| ALL RECOMMENDATIONS: HIV RESISTANCE ASSAYS |
Determining HIV Drug Resistance
|
Abbreviations: ART, antiretroviral therapy; CAB/RPV LA, injectable long-acting cabotegravir and rilpivirine. Notes:
|
Shared Decision-Making
Download Printable PDF of Shared Decision-Making Statement
Date of current publication: August 8, 2023
Lead authors: Jessica Rodrigues, MS; Jessica M. Atrio, MD, MSc; and Johanna L. Gribble, MA
Writing group: Steven M. Fine, MD, PhD; Rona M. Vail, MD; Samuel T. Merrick, MD; Asa E. Radix, MD, MPH, PhD; Christopher J. Hoffmann, MD, MPH; Charles J. Gonzalez, MD
Committee: Medical Care Criteria Committee
Date of original publication: August 8, 2023
Rationale
Throughout its guidelines, the New York State Department of Health (NYSDOH) AIDS Institute (AI) Clinical Guidelines Program recommends “shared decision-making,” an individualized process central to patient-centered care. With shared decision-making, clinicians and patients engage in meaningful dialogue to arrive at an informed, collaborative decision about a patient’s health, care, and treatment planning. The approach to shared decision-making described here applies to recommendations included in all program guidelines. The included elements are drawn from a comprehensive review of multiple sources and similar attempts to define shared decision-making, including the Institute of Medicine’s original description [Institute of Medicine 2001]. For more information, a variety of informative resources and suggested readings are included at the end of the discussion.
Benefits
The benefits to patients that have been associated with a shared decision-making approach include:
- Decreased anxiety [Niburski, et al. 2020; Stalnikowicz and Brezis 2020]
- Increased trust in clinicians [Acree, et al. 2020; Groot, et al. 2020; Stalnikowicz and Brezis 2020]
- Improved engagement in preventive care [McNulty, et al. 2022; Scalia, et al. 2022; Bertakis and Azari 2011]
- Improved treatment adherence, clinical outcomes, and satisfaction with care [Crawford, et al. 2021; Bertakis and Azari 2011; Robinson, et al. 2008]
- Increased knowledge, confidence, empowerment, and self-efficacy [Chen, et al. 2021; Coronado-Vázquez, et al. 2020; Niburski, et al. 2020]
Approach
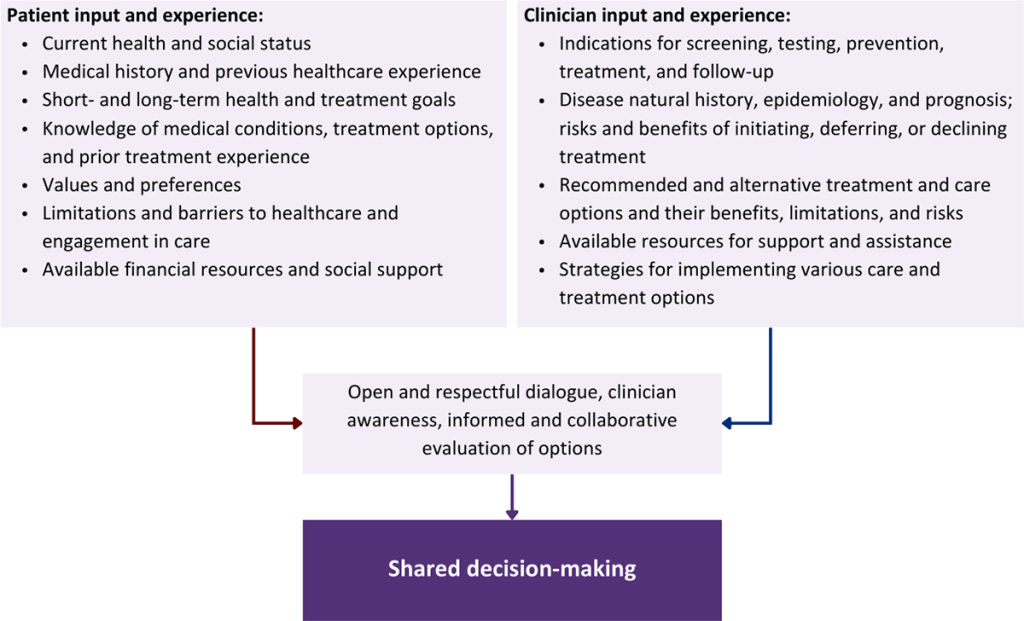
Collaborative care: Shared decision-making is an approach to healthcare delivery that respects a patient’s autonomy in responding to a clinician’s recommendations and facilitates dynamic, personalized, and collaborative care. Through this process, a clinician engages a patient in an open and respectful dialogue to elicit the patient’s knowledge, experience, healthcare goals, daily routine, lifestyle, support system, cultural and personal identity, and attitudes toward behavior, treatment, and risk. With this information and the clinician’s clinical expertise, the patient and clinician can collaborate to identify, evaluate, and choose from among available healthcare options [Coulter and Collins 2011]. This process emphasizes the importance of a patient’s values, preferences, needs, social context, and lived experience in evaluating the known benefits, risks, and limitations of a clinician’s recommendations for screening, prevention, treatment, and follow-up. As a result, shared decision-making also respects a patient’s autonomy, agency, and capacity in defining and managing their healthcare goals. Building a clinician-patient relationship rooted in shared decision-making can help clinicians engage in productive discussions with patients whose decisions may not align with optimal health outcomes. Fostering open and honest dialogue to understand a patient’s motivations while suspending judgment to reduce harm and explore alternatives is particularly vital when a patient chooses to engage in practices that may exacerbate or complicate health conditions [Halperin, et al. 2007].
Options: Implicit in the shared decision-making process is the recognition that the “right” healthcare decisions are those made by informed patients and clinicians working toward patient-centered and defined healthcare goals. When multiple options are available, shared decision-making encourages thoughtful discussion of the potential benefits and potential harms of all options, which may include doing nothing or waiting. This approach also acknowledges that efficacy may not be the most important factor in a patient’s preferences and choices [Sewell, et al. 2021].
Clinician awareness: The collaborative process of shared decision-making is enhanced by a clinician’s ability to demonstrate empathic interest in the patient, avoid stigmatizing language, employ cultural humility, recognize systemic barriers to equitable outcomes, and practice strategies of self-awareness and mitigation against implicit personal biases [Parish, et al. 2019].
Caveats: It is important for clinicians to recognize and be sensitive to the inherent power and influence they maintain throughout their interactions with patients. A clinician’s identity and community affiliations may influence their ability to navigate the shared decision-making process and develop a therapeutic alliance with the patient and may affect the treatment plan [KFF 2023; Greenwood, et al. 2020]. Furthermore, institutional policy and regional legislation, such as requirements for parental consent for gender-affirming care for transgender people or insurance coverage for sexual health care, may infringe upon a patient’s ability to access preventive- or treatment-related care [Sewell, et al. 2021].
Figure 1: Elements of Shared Decision-Making
Health equity: Adapting a shared decision-making approach that supports diverse populations is necessary to achieve more equitable and inclusive health outcomes [Castaneda-Guarderas, et al. 2016]. For instance, clinicians may need to incorporate cultural- and community-specific considerations into discussions with women, gender-diverse individuals, and young people concerning their sexual behaviors, fertility intentions, and pregnancy or lactation status. Shared decision-making offers an opportunity to build trust among marginalized and disenfranchised communities by validating their symptoms, values, and lived experience. Furthermore, it can allow for improved consistency in patient screening and assessment of prevention options and treatment plans, which can reduce the influence of social constructs and implicit bias [Castaneda-Guarderas, et al. 2016].
Clinician bias has been associated with health disparities and can have profoundly negative effects [FitzGerald and Hurst 2017; Hall, et al. 2015]. It is often challenging for clinicians to recognize and set aside personal biases and to address biases with peers and colleagues. Consciously or unconsciously, negative or stigmatizing assumptions are often made about patient characteristics, such as race, ethnicity, gender, sexual orientation, mental health, and substance use [Avery, et al. 2019; van Boekel, et al. 2013; Livingston, et al. 2012]. With its emphasis on eliciting patient information, a shared decision-making approach encourages clinicians to inquire about patients’ lived experiences rather than making assumptions and to recognize the influence of that experience in healthcare decision-making.
Stigma: Stigma may prevent individuals from seeking or receiving treatment and harm reduction services [Tsai, et al. 2019]. Among people with HIV, stigma and medical mistrust remain significant barriers to healthcare utilization, HIV diagnosis, and medication adherence and can affect disease outcomes [Turan, et al. 2017; Chambers, et al. 2015], and stigma among clinicians against people who use substances has been well-documented [Stone, et al. 2021; Tsai, et al. 2019; van Boekel, et al. 2013]. Sexual and reproductive health, including strategies to prevent HIV transmission, acquisition, and progression, may be subject to stigma, bias, social influence, and violence.
| SHARED DECISION-MAKING IN HIV CARE |
|
Resources and Suggested Reading
In addition to the references cited below, the following resources and suggested reading may be useful to clinicians.
| RESOURCES |
References
Acree ME, McNulty M, Blocker O, et al. Shared decision-making around anal cancer screening among black bisexual and gay men in the USA. Cult Health Sex 2020;22(2):201-16. [PMID: 30931831]
Avery JD, Taylor KE, Kast KA, et al. Attitudes toward individuals with mental illness and substance use disorders among resident physicians. Prim Care Companion CNS Disord 2019;21(1):18m02382. [PMID: 30620451]
Bertakis KD, Azari R. Patient-centered care is associated with decreased health care utilization. J Am Board Fam Med 2011;24(3):229-39. [PMID: 21551394]
Castaneda-Guarderas A, Glassberg J, Grudzen CR, et al. Shared decision making with vulnerable populations in the emergency department. Acad Emerg Med 2016;23(12):1410-16. [PMID: 27860022]
Chambers LA, Rueda S, Baker DN, et al. Stigma, HIV and health: a qualitative synthesis. BMC Public Health 2015;15:848. [PMID: 26334626]
Chen CH, Kang YN, Chiu PY, et al. Effectiveness of shared decision-making intervention in patients with lumbar degenerative diseases: a randomized controlled trial. Patient Educ Couns 2021;104(10):2498-2504. [PMID: 33741234]
Coronado-Vázquez V, Canet-Fajas C, Delgado-Marroquín MT, et al. Interventions to facilitate shared decision-making using decision aids with patients in primary health care: a systematic review. Medicine (Baltimore) 2020;99(32):e21389. [PMID: 32769870]
Coulter A, Collins A. Making shared decision-making a reality: no decision about me, without me. 2011. https://www.kingsfund.org.uk/sites/default/files/Making-shared-decision-making-a-reality-paper-Angela-Coulter-Alf-Collins-July-2011_0.pdf
Crawford J, Petrie K, Harvey SB. Shared decision-making and the implementation of treatment recommendations for depression. Patient Educ Couns 2021;104(8):2119-21. [PMID: 33563500]
FitzGerald C, Hurst S. Implicit bias in healthcare professionals: a systematic review. BMC Med Ethics 2017;18(1):19. [PMID: 28249596]
Greenwood BN, Hardeman RR, Huang L, et al. Physician-patient racial concordance and disparities in birthing mortality for newborns. Proc Natl Acad Sci U S A 2020;117(35):21194-21200. [PMID: 32817561]
Groot G, Waldron T, Barreno L, et al. Trust and world view in shared decision making with indigenous patients: a realist synthesis. J Eval Clin Pract 2020;26(2):503-14. [PMID: 31750600]
Hall WJ, Chapman MV, Lee KM, et al. Implicit racial/ethnic bias among health care professionals and its influence on health care outcomes: a systematic review. Am J Public Health 2015;105(12):e60-76. [PMID: 26469668]
Halperin B, Melnychuk R, Downie J, et al. When is it permissible to dismiss a family who refuses vaccines? Legal, ethical and public health perspectives. Paediatr Child Health 2007;12(10):843-45. [PMID: 19043497]
Institute of Medicine. Crossing the quality chasm: a new health system for the 21st century. 2001. https://www.ncbi.nlm.nih.gov/books/NBK222274/
KFF. Key data on health and health care by race and ethnicity. 2023 Mar 15. https://www.kff.org/racial-equity-and-health-policy/report/key-data-on-health-and-health-care-by-race-and-ethnicity/ [accessed 2023 May 19]
Livingston JD, Milne T, Fang ML, et al. The effectiveness of interventions for reducing stigma related to substance use disorders: a systematic review. Addiction 2012;107(1):39-50. [PMID: 21815959]
McNulty MC, Acree ME, Kerman J, et al. Shared decision making for HIV pre-exposure prophylaxis (PrEP) with black transgender women. Cult Health Sex 2022;24(8):1033-46. [PMID: 33983866]
Niburski K, Guadagno E, Abbasgholizadeh-Rahimi S, et al. Shared decision making in surgery: a meta-analysis of existing literature. Patient 2020;13(6):667-81. [PMID: 32880820]
Parish SJ, Hahn SR, Goldstein SW, et al. The International Society for the Study of Women’s Sexual Health process of care for the identification of sexual concerns and problems in women. Mayo Clin Proc 2019;94(5):842-56. [PMID: 30954288]
Robinson JH, Callister LC, Berry JA, et al. Patient-centered care and adherence: definitions and applications to improve outcomes. J Am Acad Nurse Pract 2008;20(12):600-607. [PMID: 19120591]
Scalia P, Durand MA, Elwyn G. Shared decision-making interventions: an overview and a meta-analysis of their impact on vaccine uptake. J Intern Med 2022;291(4):408-25. [PMID: 34700363]
Sewell WC, Solleveld P, Seidman D, et al. Patient-led decision-making for HIV preexposure prophylaxis. Curr HIV/AIDS Rep 2021;18(1):48-56. [PMID: 33417201]
Stalnikowicz R, Brezis M. Meaningful shared decision-making: complex process demanding cognitive and emotional skills. J Eval Clin Pract 2020;26(2):431-38. [PMID: 31989727]
Stone EM, Kennedy-Hendricks A, Barry CL, et al. The role of stigma in U.S. primary care physicians’ treatment of opioid use disorder. Drug Alcohol Depend 2021;221:108627. [PMID: 33621805]
Tsai AC, Kiang MV, Barnett ML, et al. Stigma as a fundamental hindrance to the United States opioid overdose crisis response. PLoS Med 2019;16(11):e1002969. [PMID: 31770387]
Turan B, Budhwani H, Fazeli PL, et al. How does stigma affect people living with HIV? The mediating roles of internalized and anticipated HIV stigma in the effects of perceived community stigma on health and psychosocial outcomes. AIDS Behav 2017;21(1):283-91. [PMID: 27272742]
van Boekel LC, Brouwers EP, van Weeghel J, et al. Stigma among health professionals towards patients with substance use disorders and its consequences for healthcare delivery: systematic review. Drug Alcohol Depend 2013;131(1-2):23-35. [PMID: 23490450]
References
Baxter J. D., Mayers D. L., Wentworth D. N., et al. A randomized study of antiretroviral management based on plasma genotypic antiretroviral resistance testing in patients failing therapy. CPCRA 046 Study Team for the Terry Beirn Community Programs for Clinical Research on AIDS. AIDS 2000;14(9):F83-93. [PMID: 10894268]
Chu C., Armenia D., Walworth C., et al. Genotypic resistance testing of HIV-1 DNA in peripheral blood mononuclear cells. Clin Microbiol Rev 2022;35(4):e0005222. [PMID: 36102816]
Cohen M. S., Shaw G. M., McMichael A. J., et al. Acute HIV-1 infection. N Engl J Med 2011;364(20):1943-54. [PMID: 21591946]
Devereux H. L., Youle M., Johnson M. A., et al. Rapid decline in detectability of HIV-1 drug resistance mutations after stopping therapy. AIDS 1999;13(18):F123-27. [PMID: 10630517]
FDA. FDA authorizes marketing of first next-generation sequencing test for detecting HIV-1 drug resistance mutations. 2019 Nov 5. https://www.fda.gov/news-events/press-announcements/fda-authorizes-marketing-first-next-generation-sequencing-test-detecting-hiv-1-drug-resistance [accessed 2023 Feb 24]
Kantor R., Gupta R. K. We should not stop considering HIV drug resistance testing at failure of first-line antiretroviral therapy. Lancet HIV 2023;10(3):e202-8. [PMID: 36610438]
McGovern R. A., Thielen A., Mo T., et al. Population-based V3 genotypic tropism assay: a retrospective analysis using screening samples from the A4001029 and MOTIVATE studies. AIDS 2010;24(16):2517-25. [PMID: 20736814]
Palella F. J., Armon C., Buchacz K., et al. The association of HIV susceptibility testing with survival among HIV-infected patients receiving antiretroviral therapy: a cohort study. Ann Intern Med 2009;151(2):73-84. [PMID: 19620160]
Vandekerckhove L., Verhofstede C., Demecheleer E., et al. Comparison of phenotypic and genotypic tropism determination in triple-class-experienced HIV patients eligible for maraviroc treatment. J Antimicrob Chemother 2011;66(2):265-72. [PMID: 21196489]
Wood B. R., Stekler J. D. Baseline HIV genotype drug resistance testing: is it time for more or less?. AIDS 2022;36(10):1449-51. [PMID: 35876702]
Updates, Authorship, and Related Guidelines
| Updates, Authorship, and Related Guidelines | |
| Date of original publication | July 13, 2016 |
| Date of current publication | June 20, 2023 |
| Highlights of changes, additions, and updates in the June 20, 2023 edition |
Comprehensive update |
| Intended users | Clinicians in New York State who provide care for patients with HIV |
| Lead author |
Steven M. Fine, MD, PhD |
| Writing group |
Rona M. Vail, MD; Joseph P. McGowan, MD, FACP, FIDSA; Samuel T. Merrick, MD; Asa E. Radix, MD, MPH, PhD; Jessica Rodrigues, MS; Charles J. Gonzalez, MD; Christopher J. Hoffmann, MD, MPH |
| Author and writing group conflict of interest disclosures | There are no author or writing group conflict of interest disclosures. |
| Committee | |
| Developer and funder |
New York State Department of Health AIDS Institute (NYSDOH AI) |
| Development process |
See Guideline Development and Recommendation Ratings Scheme, below. |
| Related NYSDOH AI guidelines |
Guidelines
Podcast |
Guideline Development and Recommendation Ratings
| Guideline Development: New York State Department of Health AIDS Institute Clinical Guidelines Program | |
| Program manager | Clinical Guidelines Program, Johns Hopkins University School of Medicine, Division of Infectious Diseases. See Program Leadership and Staff. |
| Mission | To produce and disseminate evidence-based, state-of-the-art clinical practice guidelines that establish uniform standards of care for practitioners who provide prevention or treatment of HIV, viral hepatitis, other sexually transmitted infections, and substance use disorders for adults throughout New York State in the wide array of settings in which those services are delivered. |
| Expert committees | The NYSDOH AI Medical Director invites and appoints committees of clinical and public health experts from throughout New York State to ensure that the guidelines are practical, immediately applicable, and meet the needs of care providers and stakeholders in all major regions of New York State, all relevant clinical practice settings, key New York State agencies, and community service organizations. |
| Committee structure |
|
| Disclosure and management of conflicts of interest |
|
| Evidence collection and review |
|
| Recommendation development |
|
| Review and approval process |
|
| External reviews |
|
| Update process |
|
| Recommendation Ratings Scheme | |||
| Strength | Quality of Evidence | ||
| Rating | Definition | Rating | Definition |
| A | Strong | 1 | Based on published results of at least 1 randomized clinical trial with clinical outcomes or validated laboratory endpoints. |
| B | Moderate | * | Based on either a self-evident conclusion; conclusive, published, in vitro data; or well-established practice that cannot be tested because ethics would preclude a clinical trial. |
| C | Optional | 2 | Based on published results of at least 1 well-designed, nonrandomized clinical trial or observational cohort study with long-term clinical outcomes. |
| 2† | Extrapolated from published results of well-designed studies (including nonrandomized clinical trials) conducted in populations other than those specifically addressed by a recommendation. The source(s) of the extrapolated evidence and the rationale for the extrapolation are provided in the guideline text. One example would be results of studies conducted predominantly in a subpopulation (e.g., one gender) that the committee determines to be generalizable to the population under consideration in the guideline. | ||
| 3 | Based on committee expert opinion, with rationale provided in the guideline text. | ||
Last updated on March 23, 2026